
Quantum Computing
Kvantalgoritmer kan undersöka större molekyler
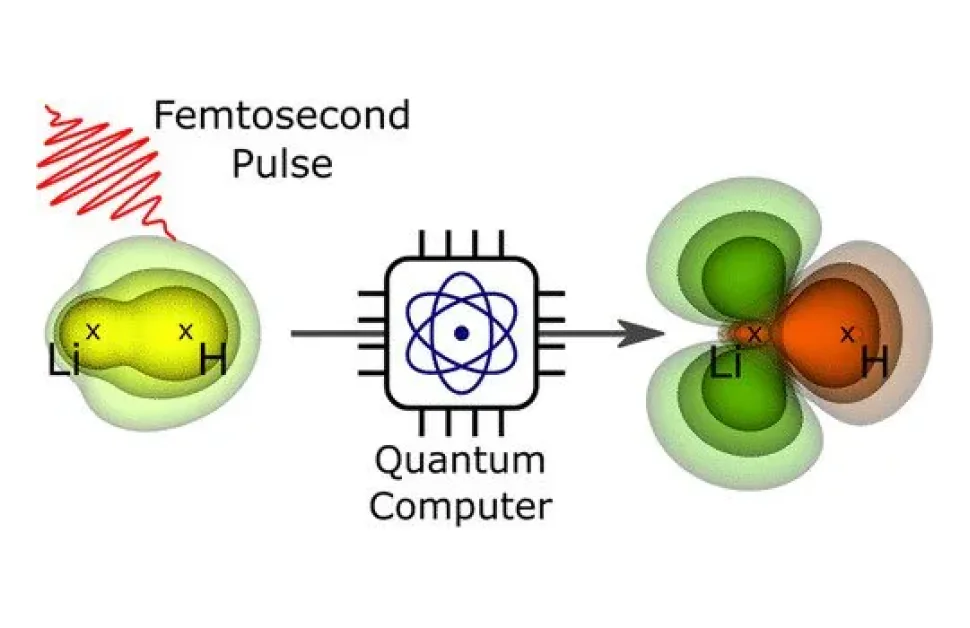
Ett team vid Helmholtz-Zentrum Berlin für Materialien und Energie (HZB) kunde beräkna elektronorbitaler och deras dynamiska utveckling på exemplet med en liten molekyl efter en laserpulsexcitation. Enligt experterna skulle denna metod kunna hjälpa till att undersöka större molekyler som inte går att beräkna med konventionella metoder.
Den nya utvecklingen hjälper till att utveckla kvantdatorer, vilket drastiskt kan minska beräkningstiden för komplexa problem.
Forskningen publicerades i Journal of Chemical Theory and Computation.
Utveckla kvantalgoritmerna
Annika Bande leder en grupp om teoretisk kemi på HZB.
"Dessa kvantdatoralgoritmer utvecklades ursprungligen i ett helt annat sammanhang. Vi använde dem här för första gången för att beräkna elektrondensiteter hos molekyler, i synnerhet även deras dynamiska utveckling efter excitation av en ljuspuls, säger Bande.
Fabian Langkabel ingår i koncernen.
"Vi utvecklade en algoritm för en fiktiv, helt felfri kvantdator och körde den på en klassisk server som simulerade en kvantdator på tio Qbits", säger Langkabel.
Teamet av forskare begränsade sin studie till mindre molekyler, vilket gjorde det möjligt för dem att utföra beräkningarna utan en riktig kvantdator. De skulle också kunna jämföra dem med konventionella beräkningar.
Fördelar jämfört med konventionella metoder
Kvantalgoritmerna ger de resultat som teamet letade efter. Till skillnad från konventionella beräkningar skulle kvantalgoritmerna kunna beräkna större molekyler med framtida kvantdatorer.
”Detta har med beräkningstiderna att göra. De ökar med antalet atomer som utgör molekylen”, fortsätter Langkabel.
När det gäller konventionella metoder multipliceras beräkningstiden med varje ytterligare atom. Men detta är inte fallet för kvantalgoritmer eftersom de blir snabbare för varje ytterligare atom.
Den nya studien visar hur man beräknar elektrondensiteter och deras "svar" på excitationer med ljus i förväg. Den använder också mycket höga rumsliga och tidsmässiga upplösningar.
Metoden gör det möjligt att simulera och förstå ultrasnabba sönderfallsprocesser, som är viktiga för kvantdatorer som består av "kvantprickar". Det gör det också möjligt att göra förutsägelser om det fysiska eller kemiska beteendet hos molekyler, vilket kan ske under absorption av ljus och överföring av elektriska laddningar.
Allt detta hjälper till att underlätta utvecklingen av fotokatalysatorer för produktion av grönt väte med solljus, och det ger bättre insikt i processerna i de ljuskänsliga receptormolekylerna i ögat.











