Calcul cuantic
Algoritmii cuantici ar putea investiga molecule mai mari
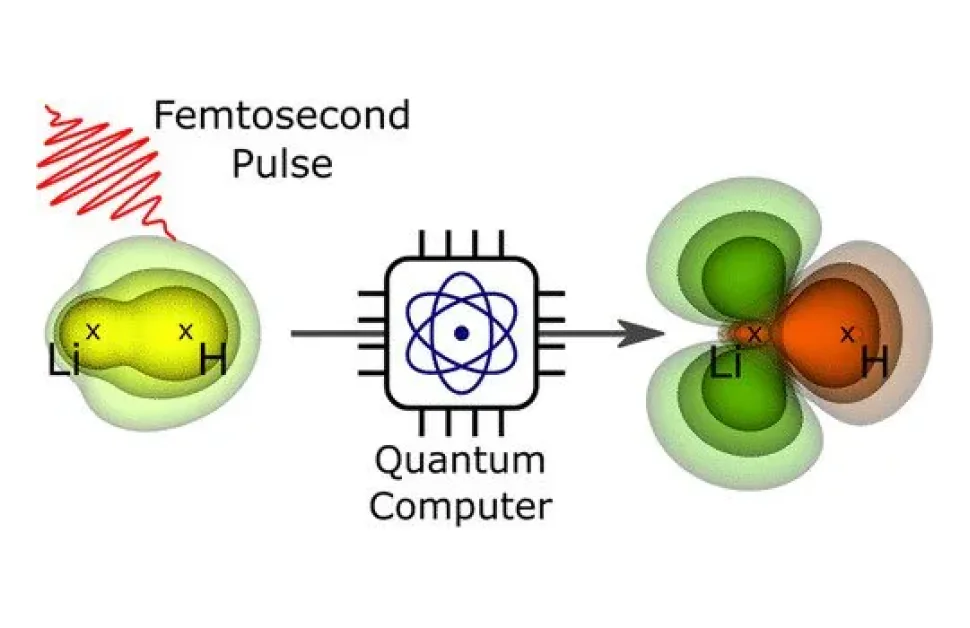
O echipă de la Helmholtz-Zentrum Berlin für Materialien und Energie (HZB) a reușit să calculeze orbitele electronice și evoluția lor dinamică pe exemplul unei molecule mici după o excitare cu puls de laser. Conform experților, această metodă ar putea ajuta la investigarea unor molecule mai mari care nu pot fi calculate cu metode convenționale.
Noua dezvoltare ajută la promovarea calculatoarelor cuantice, care ar putea reduce drastic timpul de calcul pentru probleme complexe.
Cercetarea a fost publicată în Journal of Chemical Theory and Computation.
Dezvoltarea algoritmilor cuantici
Annika Bande conduce un grup de chimie teoretică la HZB.
„Acești algoritmi de calculator cuantic au fost dezvoltați inițial într-un context complet diferit. Noi i-am folosit aici pentru prima dată pentru a calcula densitățile electronice ale moleculelor, în special și evoluția lor dinamică după excitarea cu un puls de lumină,” spune Bande.
Fabian Langkabel face parte din grup.
„Am dezvoltat un algoritm pentru un calculator cuantic fictiv, complet lipsit de erori, și l-am rulat pe un server clasic care simulează un calculator cuantic cu zece Qbit,” spune Langkabel.
Echipa de oameni de știință și-a limitat studiul la molecule mai mici, ceea ce le-a permis să efectueze calculele fără un calculator cuantic real. Ei au putut, de asemenea, să le compare cu calculele convenționale.
Avantajele față de metodele convenționale
Algoritmii cuantici produc rezultatele pe care echipa le căuta. În contrast cu calculele convenționale, algoritmii cuantici ar putea calcula molecule mai mari cu calculatoare cuantice viitoare.
„Acest lucru are legătură cu timpul de calcul. El crește odată cu numărul de atomi care alcătuiesc molecula,” continuă Langkabel.
În ceea ce privește metodele convenționale, timpul de calcul se multiplică cu fiecare atom suplimentar. Dar acest lucru nu se aplică algoritmilor cuantici, deoarece ei devin mai rapizi cu fiecare atom suplimentar.
Noua cercetare demonstrează cum să se calculeze densitățile electronice și „răspunsul” lor la excitări cu lumină în avans. Ea utilizează, de asemenea, rezoluții spațiale și temporale foarte ridicate.
Metoda face posibilă simularea și înțelegerea proceselor de decădere ultra-rapide, care sunt importante pentru calculatoarele cuantice alcătuite din „puncte cuantice.” Ea face, de asemenea, posibilă predicția comportamentului fizic sau chimic al moleculelor, care ar putea avea loc în timpul absorbției luminii și transferului de sarcini electrice.
Toate acestea ajută la facilitarea dezvoltării fotocatalizatorilor pentru producerea hidrogenului verde cu lumina soarelui și oferă o mai bună înțelegere a proceselor din moleculele receptoare sensibile la lumină din ochi.












