Kvanttilaskenta
Kvanttialgoritmit voivat tutkia suurempia molekyylejä
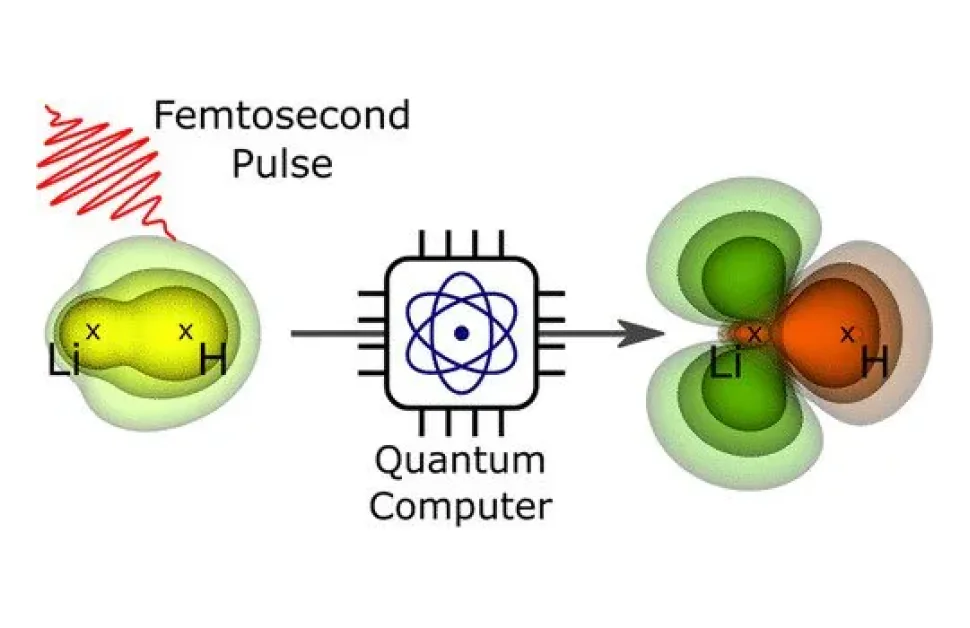
Helmholtz-Zentrum Berlin für Materialien und Energie (HZB):n tiimi pystyi laskemaan elektronien orbitaaleja ja niiden dynaamisen kehityksen pienen molekyylin esimerkissä laserpulssin herättämisen jälkeen. Asiantuntijoiden mukaan tämä menetelmä voisi auttaa tutkimalla suurempia molekyylejä, joita ei voida laskea perinteisillä menetelmillä.
Uusi kehitys edistää kvanttitietokoneita, jotka voivat leikata merkittävästi laskentaaikoja monimutkaisiin ongelmiin.
Tutkimus julkaistiin Journal of Chemical Theory and Computation -julkaisussa.
Kvanttialgoritmien kehittäminen
Annika Bande johtaa teoreettisen kemian ryhmää HZB:ssä.
“Nämä kvanttitietokonealgoritmit kehitettiin alun perin täysin eri kontekstissa. Käytimme niitä tässä ensimmäisen kerran laskemaan molekyylien elektronitiheyttä, erityisesti myös niiden dynaamista kehitystä valopulssin herättämisen jälkeen”, Bande sanoo.
Fabian Langkabel on osa ryhmää.
“Kehittimme algoritmin kuvitteelliselle, täysin virheettömälle kvanttitietokoneelle ja suoritimme sen klassisella palvelimella, joka simuloiti kvanttitietokoneen kymmenellä Q-bittillä”, Langkabel sanoo.
Tutkijaryhmä rajoitti tutkimuksensa pienempiin molekyyleihin, mikä mahdollisti laskelmien suorittamisen ilman oikeaa kvanttitietokonetta. He voivat myös vertailla niitä perinteisiin laskelmiin.
Perinteisten menetelmien edut
Kvanttialgoritmit tuottavat tulokset, joita tiimi etsi. Toisin kuin perinteiset laskelmat, kvanttialgoritmit voivat laskea suurempia molekyylejä tulevaisuuden kvanttitietokoneilla.
“Tämä liittyy laskenta-aikoihin. Ne kasvavat molekyylin atomien määrän mukaan”, Langkabel jatkaa.
Perinteisissä menetelmissä laskenta-aika moninkertaistuu jokaisen lisäatomin myötä. Tämä ei kuitenkaan ole kvanttialgoritmien tapausta, sillä ne nopeutuvat jokaisen lisäatomin myötä.
Uusi tutkimus osoittaa, miten elektronitiheyksiä ja niiden “vastetta” valon herättämiseen voidaan laskea etukäteen. Se käyttää myös erittäin korkeaa paikallista ja aikaresoluutiota.
Menetelmä mahdollistaa ultra-nopeiden hajoamisprosessien simuloimisen ja ymmärtämisen, jotka ovat tärkeitä “kvanttipisteen” kvanttitietokoneille. Se myös mahdollistaa ennusteiden tekemisen molekyylien fyysisestä tai kemiallisesta käyttäytymisestä, joka voi tapahtua valon absorboitumisen ja sähkövarauksen siirtymisen aikana.
Kaikki tämä helpottaa valokatalyyttien kehittämistä vihreän vetykaasun tuottamiseksi auringonvalolla, ja se antaa paremman ymmärryksen prosesseista valonherkkien reseptorimolekyylien silmässä.












