
Quantum Computing
Kvantealgoritmer kunne undersøge større molekyler
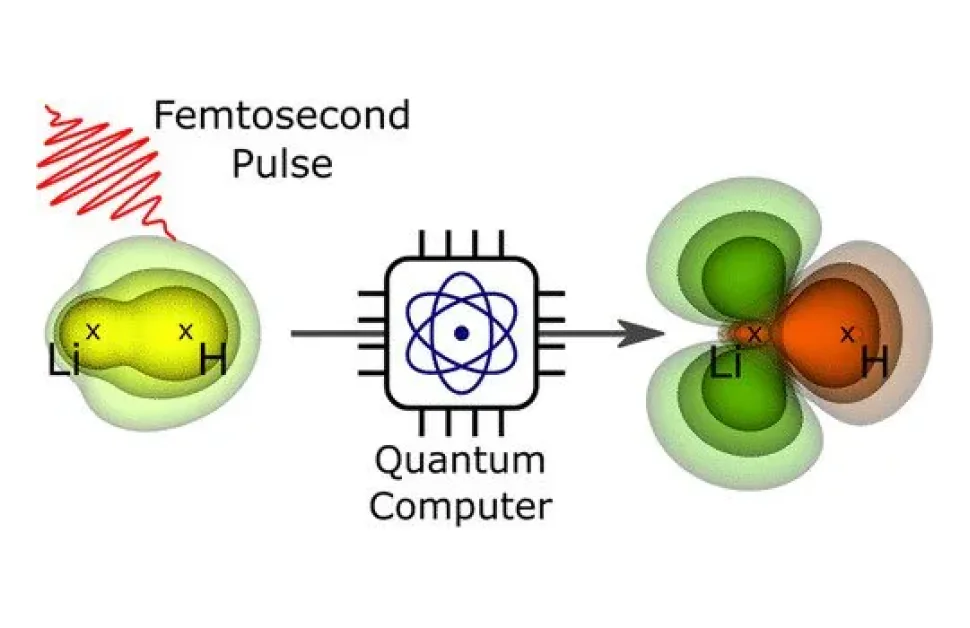
Et hold ved Helmholtz-Zentrum Berlin für Materialien und Energie (HZB) var i stand til at beregne elektronorbitalerne og deres dynamiske udvikling på eksemplet med et lille molekyle efter en laserpulsexcitation. Ifølge eksperterne kan denne metode hjælpe med at undersøge større molekyler, som ikke kan beregnes med konventionelle metoder.
Den nye udvikling hjælper med at fremme kvantecomputere, som drastisk kan reducere beregningstiden for komplekse problemer.
Forskningen blev offentliggjort i Journal of Chemical Theory and Computation.
Udvikling af kvantealgoritmer
Annika Bande leder en gruppe om teoretisk kemi på HZB.
"Disse kvantecomputeralgoritmer blev oprindeligt udviklet i en helt anden sammenhæng. Vi brugte dem her for første gang til at beregne elektrondensiteter af molekyler, især også deres dynamiske udvikling efter excitation med en lysimpuls,” siger Bande.
Fabian Langkabel er en del af koncernen.
"Vi udviklede en algoritme til en fiktiv, fuldstændig fejlfri kvantecomputer og kørte den på en klassisk server, der simulerede en kvantecomputer på ti Qbits," siger Langkabel.
Holdet af videnskabsmænd begrænsede deres undersøgelse til mindre molekyler, hvilket gjorde det muligt for dem at udføre beregningerne uden en rigtig kvantecomputer. De kunne også sammenligne dem med konventionelle beregninger.
Fordele i forhold til konventionelle metoder
Kvantealgoritmerne producerer de resultater, holdet ledte efter. I modsætning til konventionelle beregninger kunne kvantealgoritmerne beregne større molekyler med fremtidige kvantecomputere.
»Det har med beregningstiderne at gøre. De stiger med antallet af atomer, der udgør molekylet,” fortsætter Langkabel.
Når det kommer til konventionelle metoder, multipliceres regnetiden med hvert ekstra atom. Men dette er ikke tilfældet for kvantealgoritmer, da de bliver hurtigere for hvert ekstra atom.
Den nye undersøgelse demonstrerer, hvordan man på forhånd beregner elektrontætheder og deres "respons" på excitationer med lys. Den bruger også meget høje rumlige og tidsmæssige opløsninger.
Metoden gør det muligt at simulere og forstå ultrahurtige henfaldsprocesser, som er vigtige for kvantecomputere bestående af "kvanteprikker". Det gør det også muligt at komme med forudsigelser om molekylers fysiske eller kemiske adfærd, som kan finde sted under absorption af lys og overførsel af elektriske ladninger.
Alt dette er med til at lette udviklingen af fotokatalysatorer til fremstilling af grøn brint med sollys, og det giver bedre indsigt i processerne i de lysfølsomme receptormolekyler i øjet.











